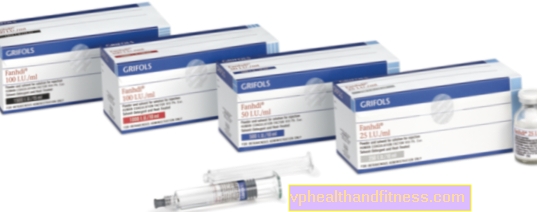
1 injectieflacon bevat 250 IE, 500 IE of 1000 IU menselijke bloedstollingsfactor VIII (FVIII) en respectievelijk 300 IE, 600 IE of 1.200 IU de menselijke von Willebrand-factor. De specifieke activiteit is minimaal 2,5 IU. tot 10 IU FVIII: C / mg eiwit afhankelijk van de verpakkingsgrootte (250 IE, 500 IE en 1000 IE). Geproduceerd uit het plasma van menselijke donoren.
| Naam | Inhoud van de verpakking | De werkzame stof | Prijs 100% | Laatst gewijzigd |
| FANHDI | 1 injectieflacon + 1 ampère spuit, poeder en reconstitutie voor bereiding oplossing voor shock en / of inf. | Factor VIII, Factor von Willebrand | 2019-04-05 |
Actie
Combinatie van stollingsfactoren - factor VIII en von Willebrand-factor zijn fysiologische componenten van menselijk plasma die fungeren als endogene componenten. Toediening van de von Willebrand-factor maakt de correctie mogelijk van hemostatische aandoeningen die optreden bij patiënten met een von Willebrand-factor-deficiëntie op twee niveaus: de von Willebrand-factor herstelt de adhesie van bloedplaatjes aan het vasculaire endotheel op de plaats van vasculaire schade (het hecht zowel aan het endotheel als aan het membraan van bloedplaatjes) hemostase, die zich manifesteert door een verkorting van de bloedingstijd (het effect verschijnt onmiddellijk en is grotendeels afhankelijk van het niveau van eiwitpolymerisatie); von Willebrand-factor veroorzaakt een vertraagde toename van gelijktijdige factor VIII-deficiëntie, een intraveneuze injectie van von Willebrand-factor bindt aan endogene factor VIII (die wordt gesynthetiseerd in de patiënt), waardoor deze factor wordt gestabiliseerd, waardoor de snelle afbraak ervan wordt voorkomen. Daarom herstelt toediening van zuivere von Willebrand-factor (een preparaat met een lage factor VIII-von Willebrand-factor) de factor VIII-spiegels naar normaal als secundair effect na de eerste infusie. Bij intraveneuze toediening aan patiënten met hemofilie bindt factor VIII zich aan de von Willebrand-factor in de bloedbaan van de patiënt. Actieve factor VIII (VIla) werkt als een cofactor van actieve factor IX (IXa) om de omzetting van factor X in actieve factor X (Xa) te versnellen. Actieve factor X zet protrombine om in trombine. Trombine zet vervolgens fibrinogeen om in fibrine, waardoor een stolsel ontstaat. Bij patiënten met hemofilie A blijft na een injectie ongeveer tweederde tot driekwart van de factor VIII in de circulatie. Het factor VIII-activiteitsniveau dat in plasma wordt bereikt, moet tussen 80% en 120% van de voorspelde factor VIII-activiteit liggen. Factor VIII-activiteit in plasma neemt af met een exponentiële tweefaseverdeling. In de beginfase vindt een verdeling tussen intravasculaire en andere compartimenten (lichaamsvloeistoffen) plaats met een plasma-eliminatiehalfwaardetijd van 3 tot 6 uur. In de volgende, langzamere fase, die waarschijnlijk het factor VIII-verbruik weerspiegelt, is de halfwaardetijd 8 tot 18 uur, met een gemiddelde van 15 uur. h.
Dosering
Intraveneus. De behandeling moet worden gestart en gecontroleerd door een arts die ervaring heeft met de behandeling van bloedingsstoornissen. Factor VIII-deficiëntie: De dosis en de duur van de behandeling zijn afhankelijk van de ernst van de factor VIIII-deficiëntie, de locatie en omvang van de bloeding en de klinische toestand van de patiënt. 1 IE De activiteit van factor VIII komt overeen met de hoeveelheid factor VIII in 1 ml normaal menselijk plasma. Toediening van 1 IU factor VIII / kg verhoogt de plasmafactor VIII-activiteit met ongeveer 1,7% - 2,5% van de normale activiteit, daarom wordt de vereiste dosis als volgt berekend: vereist aantal eenheden = lichaamsgewicht (kg) x gewenste factor VIII-toename (%) x 0,5 . De toe te dienen hoeveelheid en de toedieningsfrequentie moeten altijd afhangen van de klinische effectiviteit in een bepaald geval (soms kan het bijvoorbeeld, in aanwezigheid van een lage remmertiter, nodig zijn om een hogere dosis te gebruiken dan berekend volgens de formule). Bij bloeding en chirurgie mag de factor VIII-activiteit in de loop van de tijd niet dalen tot onder de volgende activiteitsniveaus (in% van normaal of IE / dl): vroege bloeding in gewrichten, spieren of mond - het vereiste factor VIII-niveau is 20-40% van normaal en het medicijn wordt elke 12-24 uur gedurende ten minste 1 dag toegediend, totdat het bloeden voorbij is (pijnverlichting) of de wond is genezen; meer ernstige bloeding in gewrichten, spieren of hematoom - het vereiste factor VIII-niveau is 30-60% van normaal, en medicijninfusies worden elke 12-24 uur gedurende 3-4 dagen of langer herhaald totdat pijnverlichting en acute functieverlies zijn verdwenen; levensbedreigende bloeding - het vereiste factor VIII-niveau is 60-100% van normaal, medicijninfusies worden elke 8-24 uur herhaald totdat de dreiging is verdwenen; kleine ingreep (inclusief tandextractie) - het vereiste factor VIII-niveau is 30-60%, het medicijn wordt elke 24 uur gedurende ten minste 1 dag toegediend, totdat de wond is genezen; grote operatie - vereiste factor VIII-spiegels zijn 80-100% (voor en na de operatie), infusies worden elke 8-24 uur herhaald totdat de wond is genezen en vervolgens gedurende ten minste 7 opeenvolgende dagen voortgezet, waarbij de factor VIII-spiegels op 30-60 worden gehouden % (IU / dL). De respons op de behandeling varieert individueel en daarom is controle van de factor VIII-spiegels essentieel, vooral bij grote operaties. Voor de profylaxe van bloedingen op de lange termijn bij ernstige hemofilie A is de gebruikelijke dosis factor VIII 20-40 IE / kg. om de 2-3 dagen kunnen kortere dosisintervallen of hogere doses nodig zijn bij jongere patiënten. Ziekte van Von Willebrand. Algemeen wordt aangenomen dat de toediening van 1 IE vWF: RCo / kg lichaamsgewicht het niveau van VWF: RCo in de bloedsomloop met 2% verhoogt. Het doel van de behandeling is om het niveau van VWF: RCo> 0,6 IE / ml (60%) en FVIII: C> 0,4 IE / ml (40%) in het plasma te verkrijgen. In de meeste gevallen wordt een dosis van 40-80 IE / kg lichaamsgewicht von Willebrand-factor en 20-40 IE / kg lichaamsgewicht FVIII: C aanbevolen voor hemostase. Patiënten met de ziekte van von Willebrand type 3 die mogelijk hogere doses nodig hebben om een adequaat niveau van de factor te behouden, hebben mogelijk een aanvangsdosis von Willebrand-factor van 80 IE / kg lichaamsgewicht nodig. De geselecteerde dosis dient elke 12-24 uur te worden toegediend. De dosering en de duur van de behandeling zijn afhankelijk van de klinische toestand van de patiënt, de locatie en de omvang van de bloeding en het niveau van zowel VWF: RCo als FVIII: C. Tijdens de periode waarin factor VIII-preparaten worden gebruikt die von Willebrand-factor bevatten, dient de behandelende arts rekening te houden met de mogelijkheid van een excessieve stijging van de FVIII: C-spiegels. Om een excessieve stijging van de FVIII: C-spiegels te voorkomen, moet een dosisverlaging en / of verlenging van het doseringsinterval of het gebruik van geneesmiddelen die vWF en een lagere hoeveelheid factor VIII bevatten, worden overwogen na 24-48 uur behandeling. Er zijn slechts beperkte gegevens uit klinische onderzoeken bij kinderen jonger dan 6 jaar beschikbaar voor de bovenstaande indicatie - niet gebruiken. Bij kinderen wordt bij de bovengenoemde indicaties titratie naar klinische effectiviteit ook uitgevoerd door de dosis te berekenen op basis van lichaamsgewicht. Het medicijn wordt toegediend als een langzame intraveneuze injectie (de toedieningssnelheid mag niet hoger zijn dan 10 ml / min).
Indicaties
Preventie en beheersing van bloedingen bij patiënten met hemofilie A (aangeboren tekort aan menselijke stollingsfactor VIII). Preventie en beheersing van bloeding (inclusief chirurgische bloeding) bij patiënten met de ziekte van von Willebrand (VWD) wanneer therapie met desmopressine (DDAVP) niet effectief of gecontra-indiceerd is. Het preparaat kan worden gebruikt bij de behandeling van verworven deficiëntie van menselijke stollingsfactor VIII.
Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor één van de hulpstoffen.
Voorzorgsmaatregelen
Allergische overgevoeligheidsreacties zijn mogelijk. Patiënten moeten worden geïnformeerd over de vroege tekenen van overgevoeligheidsreacties, waaronder huiduitslag, uitgebreide netelroos, beklemd gevoel op de borst, piepende ademhaling, hypotensie tot anafylactische shock. In het geval van dergelijke symptomen, moet de toediening van het geneesmiddel onmiddellijk worden stopgezet en moet, indien nodig, een passende behandeling worden ingesteld. Patiënten die met humane stollingsfactor VIII worden behandeld, moeten zorgvuldig worden gecontroleerd op de ontwikkeling van anti-factor VIII-remmers door middel van klinische observatie en beoordeling van laboratoriumtesten. Het risico op het ontwikkelen van remmers hangt samen met de duur van de blootstelling aan factor VIII, waarbij het risico het hoogst is tijdens de eerste 20 blootstellingsdagen. In zeldzame gevallen kunnen remmers zich ook ontwikkelen na de eerste 100 dagen van blootstelling. Na overschakeling op een ander factor VIII-product is terugkerende (lage titer) remmervorming waargenomen bij patiënten die eerder waren behandeld gedurende ten minste 100 dagen met een voorgeschiedenis van remmervorming. Daarom wordt aanbevolen patiënten zorgvuldig te controleren op het voorkomen van remmers na elke overschakeling op het geneesmiddel. Er bestaat een risico op trombotische complicaties bij het gebruik van geneesmiddelen met von Willebrand-factor, vooral bij patiënten met bekende klinische of laboratoriumrisicofactoren. Om deze reden is monitoring van risicopatiënten noodzakelijk om vroege tekenen van trombose op te sporen. Bovendien moeten de aanbevelingen van de huidige richtlijnen voor de behandeling en preventie van veneuze trombo-embolie worden gevolgd. Tijdens de behandelingsperiode met een geneesmiddel dat factor VIII en von Willebrand-factor bevat, dient de arts er rekening mee te houden dat langdurige behandeling een excessieve stijging van de FVIII: C-spiegels kan veroorzaken. Bij patiënten die worden behandeld met geneesmiddelen die factor VIII en von Willebrand-factor bevatten, moeten de FVIII: C-spiegels worden gecontroleerd om te voorkomen dat er te hoge spiegels blijven bestaan die het risico op trombotische complicaties kunnen verhogen. Patiënten met de ziekte van von Willebrand, vooral type 3, kunnen neutraliserende antilichamen (remmers) tegen de von Willebrand-factor ontwikkelen. Als de verwachte plasma-VWF: RCo-activiteitsniveaus niet worden bereikt, of als de bloeding onder controle wordt gehouden met geschikte doses, moet een onderzoek worden uitgevoerd om te controleren op de aanwezigheid van een von Willebrand-factor-remmer. Bij patiënten met hoge concentraties remmers is het mogelijk dat von Willbrand-factortherapie niet effectief is en dat andere therapeutische opties moeten worden overwogen. Als een centraal veneuze lijn nodig is, kunnen lokale infecties, bacteriëmie en kathetertrombose optreden. Passende vaccinatie (tegen hepatitis A- en B-virussen) moet worden overwogen bij patiënten die regelmatig van plasma afgeleide stollingsfactor VIII krijgen.
Ongewenste activiteit
Overgevoeligheid of allergische reacties (angio-oedeem, branderig en prikkelend gevoel op de injectieplaats, koude rillingen, paroxismale blozen, gegeneraliseerde urticaria, hoofdpijn, huiduitslag, bloeddrukdaling, slaperigheid, misselijkheid, rusteloosheid, tachycardie, beklemd gevoel op de borst, tintelingen, braken, piepende ademhaling), wat in sommige gevallen kan leiden tot ernstige anafylaxie met shock. Temperatuurstijgingen zijn zelden waargenomen. Patiënten met hemofilie A kunnen neutraliserende antilichamen (remmers) tegen factor VIII ontwikkelen. Wanneer dergelijke remmers zich ontwikkelen, wordt een onvoldoende klinische respons op de behandeling waargenomen. In zeer zeldzame gevallen kunnen patiënten met de ziekte van von Willebrand, met name de ziekte van type 3, neutraliserende antilichamen (remmers) tegen de von Willebrand-factor ontwikkelen. Als dergelijke remmers optreden, wordt een onvoldoende klinische respons op de behandeling waargenomen. Deze antilichamen kunnen nauw worden geassocieerd met een anafylactische reactie. Om deze reden moeten patiënten die anafylactische reacties ervaren, op remmers worden getest. In dergelijke gevallen wordt aanbevolen contact op te nemen met een gespecialiseerd centrum voor de behandeling van hemostatische aandoeningen. Er is een risico op trombotische complicaties wanneer het geneesmiddel wordt gebruikt bij patiënten met de ziekte van von Willebrand met bekende klinische of laboratoriumrisicofactoren.
Zwangerschap en borstvoeding
Het preparaat mag alleen tijdens zwangerschap en borstvoeding worden gebruikt als dit duidelijk nodig is (geen ervaring met het gebruik van het geneesmiddel vanwege het zeldzame voorkomen van hemofilie A bij vrouwen).
Interacties
Er zijn geen interacties bekend van het FVIII / VWF-syndroom met andere preparaten.
Het preparaat bevat de stof: Factor VIII, Factor von Willebrand
Geneesmiddel vergoed: NEE





















---ostatnia-deska-ratunku-gdy-inne-metody-leczenia-niepodnoci-zawodz.jpg)
