Spongiforme encefalopathieën (prionziekten) zijn die ziekten waarbij pathologische vormen van prioneiwitten bij de ontwikkeling betrokken zijn. We weten steeds meer over prionziekten, maar de belangrijkste aspecten zijn nog onbekend - de geneeskunde heeft momenteel niet de middelen om patiënten van deze ziekten te genezen.
Spongiforme encefalopathieën, d.w.z. prionziekten, kunnen zich tijdens het leven ontwikkelen, terwijl andere voortkomen uit erfelijke genmutaties die vanaf de geboorte aanwezig zijn. Binnen deze groep zijn er verschillende entiteiten die bij mensen voorkomen, voorbeelden zijn de ziekte van Creutzfeldt-Jakob of fatale familiale slapeloosheid.
Prionziekten zijn lange tijd erg mysterieus geweest. In tegenstelling tot andere ziekteverwekkers, zoals bacteriën, virussen of schimmels, bevatten ze geen nucleïnezuur - prionen worden alleen gemaakt van eiwitten. De theorie van prionziekten werd ontdekt door S. Prusiner, deze ontdekking werd zeer gewaardeerd in de wetenschappelijke gemeenschap - in 1997 ontving de onderzoeker de Nobelprijs voor de geneeskunde. Hoewel er relatief veel jaren zijn verstreken sinds het prionconcept werd geboren, geloven sommige wetenschappers nog steeds dat het onvolledig is en onderzoeken ze de aard van deze aandoeningen verder - enkele van de factoren die verantwoordelijk zijn voor spongiforme encefalopathieën zijn nu bevestigd.
Prionziekten: oorzaken
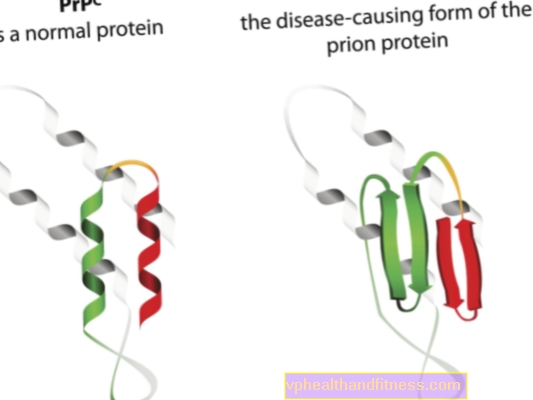
De etiologie van prionziekten houdt verband met de transformatie van normale prioneiwitten in pathogene, pathogene vormen. Prionen zijn eiwitmoleculen die in het lichaam van ieder mens worden aangetroffen. Hun functie is nog niet helemaal duidelijk, maar het is bekend dat prioneiwitten onder normale omstandigheden geen schade toebrengen aan het lichaam. Wanneer prionen echter hun structuur veranderen en pathogene deeltjes worden, ontwikkelt zich een van de vele spongiforme encefalopathieën. Prionen die van nature in het lichaam voorkomen, worden PRPC genoemd, terwijl abnormale vormen PRPSC worden genoemd. Deze laatste vormen niet alleen een ernstig probleem omdat ze zich kunnen ophopen in het zenuwweefsel in de vorm van afzettingen en schade kunnen veroorzaken, maar ook omdat ze het vermogen hebben om normale prionen om te zetten in een misvormde vorm (simpel gezegd, PRPSC normale eiwitten kunnen "infecteren" met hun pathogene potentieel).
Lees ook: Ziekte van Huntington (chorea van Huntington): oorzaken, symptomen, behandeling Spiertrillingen - oorzaken. Wat betekent spiertremor? Ziekten die het snelst doden: SCHOK, EBOLA, DAMN, AANVAL, NOODGEVALLEN [GALE ...In principe zijn er 3 oorzaken van spongiforme encefalopathie:
- sporadisch (pathogene mutatie treedt op in somatische cellen, het treedt op tijdens het leven van de patiënt),
- familie (als gevolg van de last van mutaties geërfd van ouders),
- Passage (gerelateerd aan de introductie van pathogene prionen in het menselijk lichaam, bijv. Door middel van groeihormoonpreparaten die met deze deeltjes besmet zijn of hoornvliestransplantatie van een persoon die lijdt aan een of andere spongiforme encefalopathie).
Spongiforme encefalopathieën: ziekte van Creutzfeldt-Jakob
De ziekte van Creutzfeldt-Jakob (CJD) werd voor het eerst beschreven in de vroege jaren 1920. Er zijn 4 soorten ziekten:
- sporadische CJD (de meest voorkomende, goed voor maximaal 9/10 van alle CJD-gevallen)
- geboorteplaats van CJD
- overweldigd door CJD
- variant van CJD
Het klinische beeld van verschillende varianten van de ziekte van Creutzfeldt-Jakob is wisselend. De meest voorkomende aandoeningen bij deze groep van spongiforme encefalopathieën zijn:
- dementiestoornissen (inclusief progressieve verslechtering van geheugen, aandacht en concentratie)
- myoclonus (onwillekeurige bewegingen zoals plotselinge spiertrekkingen)
- cerebellaire disfunctie (manifesteert zich bijvoorbeeld door evenwichtsstoornissen)
- wazig zicht
- piramidale en extrapiramidale symptomen
In de loop van CJD-varianten kunnen ook psychische stoornissen (bijv. Angst, depressieve stemming), pijn en andere onvrijwillige bewegingen anders dan hierboven vermeld optreden.
De prognose voor de ziekte van Creutzfeldt-Jakob is slecht - bij patiënten met sporadische CJD duurt het bijvoorbeeld gemiddeld vier tot vijf maanden vanaf het begin van de ziektesymptomen tot de dood.
Spongiforme encefalopathieën: Gerstmann-Straussler-Scheinker-syndroom
Het Gerstmann-Straussler-Scheinker-syndroom (GSS) komt meestal voor in families en wordt veroorzaakt door een erfelijke mutatie in het PRNP-gen. Het wordt beschouwd als de langzaamst voortschrijdende spongiforme encefalopathie. Het GSS-team omvat:
- spinocerebellaire ataxie
- dysartrie
- dementie stoornissen
- slikstoornissen
- nystagmus
- verhoogde spierspanning
Patiënten bij wie GSS is vastgesteld, hebben een variabele tijdsduur en bij sommige patiënten treedt de dood meer dan 10 jaar na aanvang op.
Spongiforme encefalopathieën: fatale familiale slapeloosheid
Dodelijke familiale slapeloosheid is een prionziekte die wordt veroorzaakt door mutaties in het PRNP-gen. De ziekte is uiterst zeldzaam en is tot dusver in 28 families wereldwijd gediagnosticeerd. Tijdens fatale familiale slapeloosheid is het eerste symptoom niet kunnen slapen. Dit probleem leidt tot angststoornissen en de patiënt ervaart hallucinaties. Het effect van het constante gebrek aan nachtrust zijn verstoringen in het functioneren van het autonome systeem (inclusief veranderingen in de hartfunctie, zweten en spijsverteringsstoornissen), er is ook een geleidelijke afname van het lichaamsgewicht. In meer gevorderde stadia van fatale familiaire slapeloosheid treden hormonale stoornissen op en treden symptomen van dementie op in de loop van de ziekte.
De prognose voor fatale familiaire slapeloosheid, zoals voor andere spongiforme encefalopathieën, is slecht: patiënten overlijden gewoonlijk binnen drie jaar na aanvang.
Spongiforme encefalopathieën: prionopathie met variabele gevoeligheid voor protease
Het voorkomen van deze spongiforme encefalopathieën is voornamelijk gerelateerd aan mutaties in het PRNP-gen. Deze mutaties hebben echter betrekking op verschillende codons van dit gen, en daarom worden verschillende prionziekten onderscheiden. Een relatief recent beschreven (in 2008) eenheid is prionopathie met variabele gevoeligheid voor protease. Mensen die aan deze ziekte lijden, dragen mutaties in maar liefst drie codons van het PRNP-gen.
Bij prionopathie met variabele gevoeligheid voor protease ervaren patiënten:
- cognitieve beperking
- extreme ernst van psychiatrische stoornissen: ze kunnen euforie en agitatie zijn, maar ook aanzienlijke apathie
- dysartrie
- afasie (stoornissen van taalfuncties)
De gemiddelde duur van de ziekte bij deze prionopathie is minder dan 4 jaar.
Spongiforme encefalopathieën: kuru
Kuru wordt nu beschouwd als een ziekte die praktisch niet meer bestaat - het werd gevonden bij vertegenwoordigers van stammen uit Papoea-Nieuw-Guinea, die kannibalistisch gedrag vertoonden. Het dominante symptoom van deze spongiforme encefalopathie is progressieve cerebellaire ataxie. Het kan gepaard gaan met onvrijwillige bewegingen (voornamelijk in de vorm van chorea, tremoren en athetose), evenals urine- en fecale incontinentie. Patiënten op kuru ervaren ook aanzienlijke stemmingswisselingen, ze ontwikkelen primitieve reflexen (bijvoorbeeld zuigen). Een heel kenmerkend probleem in het geval van deze prionziekte zijn gedwongen huil- of lachbuien - vanwege het laatste fenomeen wordt kuru soms de "lachende dood" genoemd.
Spongiforme encefalopathieën: diagnose
Prionziekten kunnen worden vermoed op basis van de symptomen van de patiënt. Ze zijn echter vrij niet-specifiek, omdat ze ook kunnen voorkomen in de loop van een aantal andere ziekten die geen verband houden met prionen. Om deze reden worden de volgende ook gebruikt bij de diagnose van spongiforme encefalopathieën:
- beeldvormingstests (bv. magnetische resonantiebeeldvorming, waarmee veranderingen kunnen worden gedetecteerd die verband houden met de degeneratie van de hersenen door prioneiwitten)
- laboratoriumtesten (zoals de bepaling van eiwitconcentraties in het cerebrospinale vocht, bijv.MAP-tau, S-100 of 14-3-3 eiwitten),
- genetische tests (om de aanwezigheid van mutaties bij de patiënt te detecteren),
- immunhistochemische tests (met antilichamen tegen prioneiwitten).
De diagnose kan ook worden bevestigd door autopsie van de hersenen, waarbij het mogelijk is veranderingen te vinden die kenmerkend zijn voor spongiforme encefalopathieën. Dit kunnen sponsachtige laesies zijn, verschillend verspreid en met een verschillende structuur (afhankelijk van de specifieke ziekte-entiteit), amyloïde plaques en neuronale defecten.
Spongiforme encefalopathieën: behandeling
Prionziekten zijn momenteel ongeneeslijk - ondanks talloze onderzoeken die al vele jaren aan de gang zijn, heeft de geneeskunde nog steeds geen medicijnen die hun vooruitgang kunnen vertragen of volledig kunnen remmen. Symptomatische behandeling wordt toegepast bij patiënten met spongiforme encefalopathieën, die erop gericht is de intensiteit van de symptomen te verlichten en hun kwaliteit van leven zoveel mogelijk te verbeteren.
Er wordt echter nog gewerkt aan de behandeling van spongiforme encefalopathieën. Wetenschappers proberen verschillende methoden te gebruiken - het eerste voorbeeld is gentherapie. Ze zouden nucleïnezuren en de mutaties in hun structuur aantasten - het doel van gentherapie zou zijn om fouten in de genetische code te neutraliseren. Een andere benadering is de basis van immuuntherapie - er wordt gewerkt aan het maken van antilichamen waarvan de rol zou zijn om pathogene prionen te elimineren. Een andere methode die het potentieel ziet om spongiforme encefalopathieën te bestrijden, is de behandeling met behulp van gesynthetiseerde eiwitmoleculen, die, eenmaal geïntroduceerd in het lichaam van de patiënt, pathologische eiwitten neutraliseren.
Aanbevolen artikel:
Encefalopathieën - oorzaken, typen en symptomen