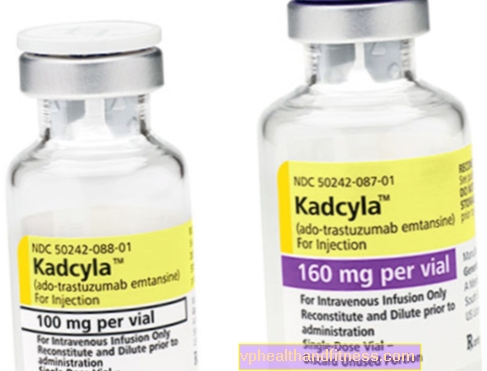
1 injectieflacon bevat 100 mg (160 mg) poeder voor reconstitutie 5 ml (8 ml) trastuzumab-emtansineconcentraat in een concentratie van 20 mg / ml voor infusie na reconstitutie.
| Naam | Inhoud van de verpakking | De werkzame stof | Prijs 100% | Laatst gewijzigd |
| Kadcyla | 1 injectieflacon, poeder voor bereiding Uiteindelijke oplossing naar inf. | Trastuzumab-emtansine | 2019-04-05 |
Actie
Geneesmiddel tegen kanker. Trastuzumab-emtansine is een antilichaam-geneesmiddelconjugaat dat trastuzumab bevat, een gehumaniseerd anti-HER2 IgG1 monoklonaal antilichaam, covalent gekoppeld aan DM1 (maytansinederivaat, microtubuli-remmer) via een stabiele MCC-thioetherlink (4- cyclohexylaat-1-carboxylaat) . Emtansine is het MCC-DM1-complex. De conjugatie van DM1 met trastuzumab veroorzaakt de selectieve werking van cytotoxische geneesmiddelen tegen tumorcellen die HER2 tot overexpressie brengen en verhoogt zo de intracellulaire concentratie van DM1 rechtstreeks in de tumorcellen. Bij binding aan HER2 wordt trastuzumab-emtansine receptorgemedieerd geïnternaliseerd, gevolgd door lysosomale afbraak, waarbij DM1-bevattende katabolieten vrijkomen (voornamelijk lysine-MCC-DM1). Het werkingsmechanisme van trastuzumab-emtansine wordt gemedieerd door de activiteit van trastuzumab en DM1. Trastuzumab-emtansine bindt, net als trastuzumab, aan domein IV van het extracellulaire domein (ECD) van de receptor, evenals aan Fcγ-receptoren en complementeert C1q. Bovendien remt het de activiteit van het ECD-domein van de HER2-receptor, remt het de signalering van de fosfatidylinositol 3-kinase (PI3-K) -route en bemiddelt het antilichaamafhankelijke cellulaire cytotoxiciteit (ADCC) in menselijke borstkankercellen die HER2 tot overexpressie brengen. DM1 bindt aan tubuline. Door de tubulinepolymerisatie te remmen, zorgen zowel DM1 als trastuzumab-emtansine ervoor dat cellen in de G2 / M-fase van de celcyclus tot stilstand komen, wat uiteindelijk leidt tot celdood door apoptose. De MCC-linker vermindert de systemische afgifte van DM1 en verhoogt de concentratie op de doellocatie. Trastuzumab-emtansine wordt gedeconjugeerd en vervolgens door proteolyse in de cellysosomen gekataboliseerd. DM1 wordt voornamelijk gemetaboliseerd door CYP3A4 en in mindere mate door CYP3A5. De T0.5 van trastuzumab is ongeveer 4 dagen. Er was geen accumulatie van trastuzumab-emtansine na meerdere intraveneuze infusies die met een tussenpoos van 3 weken werden gegeven. Leeftijd had geen effect op de farmacokinetiek van trastuzumab-emtansine.
Dosering
Intraveneus. Het preparaat moet worden voorgeschreven door een arts en worden toegediend onder toezicht van professionele zorgverleners die ervaring hebben met de behandeling van kankerpatiënten. Patiënten die trastuzumab-emtansine krijgen, moeten een HER2-positieve kanker-immunohistochemie (IHC) -score 3+ of een ratio ≥2 bij in situ hybridisatie (ISH) hebben. Tests moeten worden uitgevoerd met behulp van in vitro diagnostische (IVD) tests met CE-markering. Als de CE IVD-test niet beschikbaar is, moet de test worden uitgevoerd met een andere gevalideerde test. Om medische fouten te voorkomen, is het belangrijk om het etiket van de injectieflacon te controleren om er zeker van te zijn dat het geneesmiddel dat wordt bereid en toegediend, Kadcyla (trastuzumab-emtansine) is en niet Herceptin (trastuzumab). De aanbevolen dosis is 3,6 mg / kg lichaamsgewicht. gegeven als intraveneuze infusie om de 3 weken (cyclus van 21 dagen). Patiënten moeten worden behandeld tot tumorprogressie of het bereiken van onaanvaardbare toxiciteit. De startdosis moet worden toegediend als een intraveneuze infusie van 90 minuten. De injectieplaats moet nauwlettend worden gecontroleerd, aangezien het geneesmiddel tijdens toediening subcutaan kan doordringen. Als de vorige infusie goed werd verdragen, kunnen volgende doses worden gegeven als een infuus van 30 minuten. Patiënten moeten worden geobserveerd tijdens de infusie en gedurende ten minste 30 minuten daarna. De infusiesnelheid moet worden verlaagd of onderbroken als de patiënt symptomen ervaart die verband houden met de infusie. Trastuzumab-emtansine moet worden stopgezet in geval van levensbedreigende infusiereacties. Medicijnen voor allergische / anafylactische infusiereacties en noodapparatuur moeten beschikbaar zijn voor onmiddellijk gebruik. Als een geplande dosis wordt gemist, moet deze zo snel mogelijk worden toegediend; wacht niet tot de volgende cyclus. Het doseringsschema moet worden aangepast om het doseringsinterval van 3 weken te behouden. De volgende dosis moet worden toegediend volgens de doseringsaanbevelingen. De behandeling van symptomatische bijwerkingen kan bestaan uit periodieke stopzetting, dosisverlaging of stopzetting van de behandeling. Schema voor dosisverlaging (startdosis 3,6 mg / kg): eerste dosisverlaging 3 mg / kg; tweede dosisverlaging 2,4 mg / kg lichaamsgewicht; als verdere dosisverlaging nodig is, moet de behandeling worden stopgezet. Richtlijnen voor dosisaanpassing voor ASAT- en ALAT-verhogingen: Graad 2 (> 2,5 tot ≤5 x ULN) - geen dosisaanpassing nodig; graad 3 (> 5 tot ≤20 x ULN) - gebruik het preparaat als AST / ALT daalt tot graad ≤ 2 (> 2,5 tot 20 x ULN) - stop de behandeling. Dosisaanpassingsregels voor hyperbilirubinemie: graad 2 (> 1,5 tot ≤3 x ULN) - gebruik het preparaat wanneer het bilirubinespiegel is gedaald tot graad ≤1. (> ULN tot 1,5 x ULN), geen dosisaanpassing nodig; graad 3 (> 3 tot ≤10 x ULN) - gebruik het preparaat als de bilirubineconcentratie daalt tot ≤ graad 1 (> ULN tot 1,5 x ULN), verlaag vervolgens de dosis (zie schema voor dosisverlaging); Graad 4 (> 10 x ULN) - Beëindiging behandeling. Dosisaanpassingsregels voor trombocytopenie: graad 3 (bloedplaatjesconcentratie 25.000 tot 3) - gebruik het preparaat wanneer de bloedplaatjesconcentratie ≤1 is. (bijv.> 75.000 / mm3); geen dosisaanpassing nodig; Graad 4 (bloedplaatjesconcentratie 3) - gebruik het preparaat wanneer de bloedplaatjesconcentratie ≤1 bereikt. (bijv. ≥75.000 / mm3) en verlaag vervolgens de dosis (zie schema voor dosisverlaging). Principes voor dosisaanpassing bij patiënten met ventriculaire disfunctie: LVEF 45% - voortzetting van de therapie; LVEF 40% tot ≤45% met gelijktijdige reductie van de ejectiefractie door - gebruik het preparaat niet; Beoordeel de LVEF opnieuw binnen 3 weken; stop de behandeling als de LVEF nog steeds ≥ 10 procentpunten ten opzichte van de uitgangswaarde is; symptomatische CHF - stop de behandeling. Kinderen en adolescenten: De veiligheid en werkzaamheid bij kinderen en adolescenten jonger dan 18 jaar zijn niet vastgesteld, aangezien uitgezaaide borstkanker (MBC) bij deze populatie niet bekend is. Speciale patiëntengroepen. De behandeling moet tijdelijk worden onderbroken bij patiënten die graad 3 of 4 perifere neuropathie ontwikkelen tot graad ≤ 2; wanneer de behandeling opnieuw wordt gestart, kan worden overwogen de dosis te verlagen zoals beschreven in het schema voor dosisverlaging. Er is geen dosisaanpassing nodig voor patiënten ≥ 65 jaar; er zijn onvoldoende gegevens om de veiligheid en werkzaamheid van therapie bij patiënten ≥75 jaar te bepalen. Er is geen aanpassing van de aanvangsdosis nodig bij patiënten met een lichte of matige nierfunctiestoornis; De mogelijke noodzaak van dosisaanpassing bij patiënten met ernstige nierinsufficiëntie kan niet worden bepaald en daarom moeten deze patiënten zorgvuldig worden gecontroleerd. Er is geen aanpassing van de aanvangsdosis nodig bij patiënten met een lichte of matige leverfunctiestoornis. Trastuzumab-emtansine is niet onderzocht bij patiënten met een ernstige leverfunctiestoornis; Voorzichtigheid is geboden bij de behandeling van patiënten met leverinsufficiëntie vanwege bekende hepatotoxiciteit waargenomen tijdens de behandeling. Manier van geven. Het preparaat moet door medisch personeel worden opgelost en verdund en als intraveneuze infusie worden toegediend. Het mag niet als bolus of snelle injectie worden gegeven.
Indicaties
Monotherapie van volwassen patiënten met HER2-positieve, inoperabele lokaal gevorderde of gemetastaseerde borstkanker, eerder behandeld met trastuzumab en een taxaan, in combinatie of afzonderlijk. Patiënten die eerder zijn behandeld voor lokaal gevorderde of gegeneraliseerde ziekte, of die een recidief hebben ondergaan tijdens of binnen 6 maanden na voltooiing van adjuvante therapie.
Contra-indicaties
Overgevoeligheid voor de werkzame stof of andere ingrediënten van het preparaat.
Voorzorgsmaatregelen
Gevallen van interstitiële longziekte (ILD), waaronder pneumonie, zijn gemeld in klinische onderzoeken met trastuzumab-emtansine; sommige zijn in verband gebracht met acuut respiratoir falen of waren fataal. Stopzetting van de behandeling met het preparaat wordt aanbevolen bij patiënten met de diagnose ILD of pneumonie. Patiënten met dyspneu in rust, geassocieerd met complicaties van gevorderde kanker en daarmee samenhangende ziekten, kunnen een verhoogd risico lopen op het ontwikkelen van pulmonale complicaties. Tijdens de behandeling met het preparaat zijn hepatotoxiciteit en ernstige aandoeningen van de lever en galwegen waargenomen, waaronder nodulaire regeneratieve hypertrofie (NRH) van de lever en sterfgevallen als gevolg van door geneesmiddelen geïnduceerd leverletsel; comorbiditeit en / of gelijktijdige medicatie waarvan bekend is dat ze hepatotoxisch zijn, kunnen ook zijn aangetast. De leverfunctie moet worden gecontroleerd voordat de behandeling wordt gestart en voorafgaand aan de daaropvolgende dosering. Patiënten met een ALAT-verhoging bij aanvang (bijv. Geassocieerd met levermetastasen) zijn vatbaar voor de ontwikkeling van leverfalen en lopen een hoger risico op graad 3-5 levertoxiciteit of verhoogde laboratoriumwaarden in de lever. Gevallen van NRH werden gediagnosticeerd door het nemen van leverbiopsieën. Het optreden van NRH moet in overweging worden genomen bij alle patiënten met klinische symptomen van portale hypertensie en / of cirrose-achtige beelden die te zien zijn op computertomografie van de lever, maar met normale serumtransaminasewaarden en geen ander bewijs van cirrose. Als NRH wordt gediagnosticeerd, moet de behandeling met het preparaat worden stopgezet. Het is niet onderzocht bij patiënten met serumtransaminasen> 2,5 x ULN (bovengrens van normaal) of met totaal bilirubine> 1,5 x ULN voorafgaand aan de start van de behandeling. In het geval van serumtransaminaseactiviteit> 3 x ULN en gelijktijdig een totale bilirubineconcentratie> 2 x ULN, moet de behandeling worden beëindigd. Voorzichtigheid is geboden bij de behandeling van patiënten met leverinsufficiëntie. Er is een verhoogd risico op linkerventrikeldisfunctie tijdens de behandeling. Bij patiënten die werden behandeld met trastuzumab-emtansine, werd een afname van de linkerventrikelejectiefractie (LVEF) na 50 jaar) waargenomen, met een aanvankelijke lage LVEF-waarde (25 kg / m2). Standaard hartfunctietesten (echocardiogram of multi-gate radionuclide-angiografie) dienen voorafgaand aan de start van de behandeling en met regelmatige tussenpozen (bijv. Elke 3 maanden) te worden uitgevoerd. De LVEF van de patiënten bij baseline was ≥50% in de meeste klinische onderzoeken. Patiënten met congestief hartfalen, ernstige aritmieën die behandeling behoeven, een voorgeschiedenis van myocardinfarct of instabiele angina pectoris binnen 6 maanden voorafgaand aan randomisatie, of met dyspneu in rust als gevolg van gevorderde neoplastische ziekte, werden uitgesloten van het onderzoek. In het geval van linkerventrikelstoornissen moet de toediening van de volgende dosis worden uitgesteld of moet de therapie worden stopgezet. Het effect van behandeling met trastuzumab-emtansine bij patiënten die de behandeling met trastuzumab stopzetten vanwege een infusiegerelateerde reactie, is niet onderzocht; therapie met dit medicijn wordt niet aanbevolen bij deze patiëntengroep. De therapie moet worden stopgezet bij patiënten die een ernstige infusiegerelateerde reactie ontwikkelen totdat de symptomen verdwijnen. Opnieuw starten van de therapie dient te worden overwogen op basis van klinische beoordeling van de ernst van de reactie. De behandeling moet worden stopgezet in geval van een levensbedreigende infusiereactie. Het effect van behandeling met trastuzumab-emtansine bij patiënten die de behandeling met trastuzumab hebben stopgezet vanwege overgevoeligheid, is niet onderzocht; Het gebruik van trastuzumab-emtansine bij deze patiënten wordt niet aanbevolen. Patiënten moeten worden gecontroleerd op overgevoeligheids- / allergische reacties, welke symptomen hetzelfde kunnen zijn als infusiegerelateerde reacties; Er zijn ernstige anafylactische reacties waargenomen. Als zich overgevoeligheid (met verhoogde reacties tijdens volgende infusies) ontwikkelt, moet de behandeling met trastuzumab-emtansine worden stopgezet.Vanwege het risico op hemorragische voorvallen (waaronder CZS-, respiratoire en gastro-intestinale bloeding), is voorzichtigheid geboden en dient aanvullende controle te worden overwogen bij gelijktijdige toediening met anticoagulantia of plaatjesaggregatieremmers. Het wordt aanbevolen om het aantal bloedplaatjes te controleren voorafgaand aan toediening van elke dosis trastuzumab-emtansine. Patiënten met trombocytopenie (≤ 100.000 / mm3) en patiënten die anticoagulantia krijgen (bijv. Warfarine, heparine, heparines met laag molecuulgewicht) moeten tijdens de behandeling met trastuzumab-emtansine nauwlettend worden gevolgd. Trastuzumab-emtansine is niet onderzocht bij patiënten met een trombocytenaantal ≤ 100.000 / mm3 voorafgaand aan de start van de therapie. In geval van verergering van trombocytopenie tot graad 3 of hoger (3), mag het preparaat niet worden gebruikt totdat de toxiciteit afneemt tot graad 1 (≥75.000 / mm3). In klinische onderzoeken met trastuzumab-emtansine is graad 1 perifere neuropathie, voornamelijk sensorisch, opgetreden. Patiënten met perifere neuropathie van graad ≥ 3 werden uitgesloten van deelname aan klinische onderzoeken. De behandeling van patiënten met graad 3 of 4 perifere neuropathie moet periodiek worden onderbroken totdat de complicatie is verminderd tot graad ≤ 2. Patiënten moeten regelmatig worden gecontroleerd op tekenen van neurotoxiciteit. Om de traceerbaarheid van biologische preparaten te verbeteren, dient de handelsnaam van het toegediende geneesmiddel duidelijk te worden geschreven (of vermeld) in het patiëntendossier. De veiligheid en werkzaamheid van het geneesmiddel bij kinderen en adolescenten onder de 18 jaar zijn niet vastgesteld, aangezien uitgezaaide borstkanker bij deze populatie niet wordt aangetroffen. Het mag niet als bolus of snelle injectie worden gegeven.
Ongewenste activiteit
Zeer vaak: urineweginfectie, trombocytopenie, anemie, hypokaliëmie, slapeloosheid, perifere neuropathie, hoofdpijn, bloeding, epistaxis, hoesten, dyspneu, stomatitis, diarree, braken, misselijkheid, obstipatie, droge mond, buikpijn huiduitslag, musculoskeletale pijn, artralgie, spierpijn, vermoeidheid, koorts, asthenie, koude rillingen, verhoging van transaminasen. Vaak: neutropenie, leukopenie, overgevoeligheid, duizeligheid, dysgeusie, geheugenstoornis, droge-ogensyndroom, conjunctivitis, visusstoornissen, verhoogde traanproductie, linkerventrikeldisfunctie, hypertensie, dyspepsie, tandvleesbloeding, huidpruritus, alopecia, nagelziekte palmoplantaire erytrodysesthesie, urticaria, perifeer oedeem, verhoogde alkalische fosfatasespiegels, infusiegerelateerde reacties (roodheid van de huid, koude rillingen, koorts, dyspneu, lage bloeddruk, piepende ademhaling, bronchospasme, tachycardie). Soms: pneumonie (ILD), hepatotoxiciteit, leverfalen, nodulaire regeneratieve hyperplasie, portale hypertensie, extravasatie op de injectieplaats (erytheem, gevoeligheid, huidirritatie, pijn of zwelling). Bovendien is hyperbilirubinemie waargenomen. In klinische onderzoeken traden verhogingen van serumtransaminasen (graad 1-4) op tijdens behandeling met trastuzumab-emtansine en waren deze over het algemeen van voorbijgaande aard. De verhoging van transaminasen was meestal van voorbijgaande aard, met een piek op de 8e dag na toediening. Daarna nam de toxiciteit af tot graad 1 of verdween voor de volgende cyclus. Er was ook een cumulatief effect (het percentage patiënten met graad 1-2 ALAT / ASAT-verhoging nam toe met de volgende cycli). Bij patiënten met verhoogde transaminasespiegels ervoer de meerderheid van de patiënten een vermindering of herstel van de toxiciteit van graad 1 binnen 30 dagen na de laatste dosis trastuzumab-emtansine. Linkerventrikeldisfunctie werd gevonden bij 2,2% van de patiënten die aan klinische onderzoeken deelnamen. In de meeste gevallen waren de LVEF-verlagingen van graad 1 of 2 asymptomatisch en werd toxiciteit van graad 3 of 4 waargenomen bij 0,4% van de patiënten, gewoonlijk tijdens de eerste behandelingscycli (1–2). Ernstige episodes van hemorragische voorvallen (graad ≥ 3) werden gemeld bij 2,2% van alle patiënten. Trombocytopenie trad op bij 24,9% van de patiënten in klinische onderzoeken en was de meest voorkomende bijwerking die leidde tot stopzetting van de behandeling. In klinische onderzoeken waren de incidentie en ernst van trombocytopenie hoger bij Aziatische patiënten. Sommige patiënten die deze complicaties ontwikkelden, werden ook behandeld met antistolling. Er waren fatale bloedingsepisodes en ernstige bloedingscomplicaties, waaronder bloedingen in het centraal zenuwstelsel. 5,3% van de patiënten testte positief op anti-trastuzumab-emtansine-antilichamen.
Zwangerschap en borstvoeding
Het gebruik van trastuzumab-emtansine bij zwangere vrouwen wordt niet aanbevolen. Vóór de zwangerschap moeten vrouwen worden geïnformeerd over de mogelijkheid van schade aan de foetus. Patiënten die zwanger worden, moeten echter onmiddellijk contact opnemen met een arts. Als een zwangere vrouw wordt behandeld met trastuzumab-emtansine, wordt nauwlettende controle door een multidisciplinair team aanbevolen. Trastuzumab kan schade aan de foetus of de dood veroorzaken als het wordt toegediend aan een zwangere vrouw. Bij kinderen van zwangere vrouwen die met het preparaat werden behandeld, zijn gevallen van oligohydramnion gemeld, waarvan sommige tot fatale longhypoplasie leidden. DM1, de cytotoxische component van trastuzumab-emtansine, kan teratogeen en mogelijk embryotoxisch zijn. Vrouwen dienen de borstvoeding te stoppen voordat de behandeling met trastuzumab-emtansine wordt gestart. Patiënten mogen 7 maanden na stopzetting van de behandeling beginnen met borstvoeding. Vrouwen die zwanger kunnen worden, moeten effectieve anticonceptie gebruiken tijdens de behandeling met het geneesmiddel en gedurende 7 maanden na de laatste dosis trastuzumab-emtansine. Effectieve anticonceptiemethoden moeten ook worden gebruikt door mannen of hun vrouwelijke partners.
Opmerkingen
Patiënten met infusiegerelateerde reacties mogen niet rijden of machines bedienen totdat deze symptomen zijn verdwenen. Informatie opgesteld op basis van SPC van 13 juli 2017 De huidige SmPC is beschikbaar op www.roche.pl.
Interacties
In-vitro-resultaten van metabolismestudies in menselijke levermicrosomen geven aan dat DM1 voornamelijk wordt gemetaboliseerd door het enzym CYP3A4 en in mindere mate door CYP3A5. Gelijktijdig gebruik van sterke CYP3A4-remmers (bijv. Ketoconazol, itraconazol, claritromycine, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, saquinavir, telithromycine en voriconazol) moet worden vermeden, aangezien dit de DM1-spiegels en toxiciteit kan verhogen. Een alternatieve formulering die CYP3A4 niet of slechts in geringe mate remt, moet worden overwogen. Als gelijktijdig gebruik van sterke CYP3A4-remmers niet kan worden vermeden, en waar mogelijk, overweeg dan om de toediening van trastuzumab-emtansine uit te stellen totdat de CYP3A4-remmers uit de circulatie zijn geklaard (ongeveer 3 halfwaardetijden van de remmers). Als echter gelijktijdig een sterke CYP3A4-remmer wordt gebruikt en de behandeling met trastuzumab-emtansine niet kan worden uitgesteld, moet de patiënt in dergelijke gevallen nauwlettend worden gecontroleerd op bijwerkingen.
Het preparaat bevat de stof: Trastuzumab-emtansine
Geneesmiddel vergoed: NEE


.jpg)